c7c7.app第一醫院劉立新團隊在肝纖維化形成及其機制方面取得進展
肝纖維化是一種常見的病理狀態,其發病機制主要是多種病因引起肝臟發生損傷���,多種炎性細胞因子如轉化生長因子β1(Transforming growth factor beta 1,TGFβ1)等分泌增多�����,從而導致肝星狀細胞(Hepatic stellate cells���,HSCs)活化����,細胞外基質(Extracellular matrix����,ECM)合成增多�����,最終形成肝纖維化�������。肝纖維化是可逆的��������,如果得不到很好的控制�����,將導致不可逆的肝硬化����,乃至肝癌���。HSCs活化是肝纖維化發生發展的中心環節����。在正常的肝臟中�,HSCs主要位于肝臟Disse間隙���������,處于靜止狀態�������。當肝臟發生損傷時�����,靜止的HSCs被激活并轉變為肌成纖維細胞���?�;罨腍SCs遷移至組織受損部位并逐漸積累�������,不僅產生大量的ECM����������,而且可以防止ECM被降解����。因此抑制HSCs活化被認為是抑制肝纖維化的有效策略���������。越來越多的研究表明����,自噬可以促進HSCs活化��,同時自噬也可以為活化的HSCs提供能量以促使其存活��������。自噬的調控是復雜的���������,其中PI3K/AKT/mTOR信號通路是調節細胞自噬的主要通路。長鏈非編碼RNA H19被發現參與了自噬體的延伸/閉合�����,對自噬有促進作用�����,具體機制可能與PI3K/AKT/mTOR信號通路有關。
胰島素樣生長因子結合蛋白相關蛋白1(Insulin-like growth factor binding protein-associated protein 1�������,IGFBPrP1)是導師課題組十余年來研究發現的一種新的致肝纖維化因子�������。前期一系列研究表明���������,IGFBPrP1能夠活化HSCs���������,產生過多的ECM���,促進肝纖維化的發生發展����������。另外��������,IGFBPrP1和TGFβ1可以相互調節,共同促進HSCs活化���������。TGFβ1是已知的最強的致肝纖維化因子����,可通過自噬激活HSCs�������,PI3K/AKT信號通路參與了其對自噬的調控���������。TGFβ1也能促進H19的表達������,其機制可能與PI3K/AKT的活化有關������。鑒于TGFβ1與H19和自噬有關����,而IGFBPrP1與TGFβ1可互為因果���������,因此本研究以長鏈非編碼RNA H19和自噬作為切入點����,研究H19是否通過自噬調控IGFBPrP1致肝纖維化形成的作用及其機制������。
在這項研究中����,研究團隊首先建立膽總管結扎(Bile duct ligation,BDL)誘導的小鼠肝纖維化模型���������,確定肝纖維化形成過程中IGFBPrP1與H19�����、自噬的表達變化��������。結果顯示IGFBPrP1與H19、自噬呈伴隨性增高��。接著研究團隊分別將腺病毒介導的IGFBPrP1(AdIGFBPrP1)在體內通過小鼠尾靜脈注射轉染C57BL/6小鼠肝組織������,同時在體外轉染JS-1(小鼠肝星狀細胞株)��,通過qRT-PCR���������、Western blotting��������、免疫組織化學����、透射電子顯微鏡等分子生物學實驗證實,IGFBPrP1激發了自噬的表達����。為了明確自噬對IGFBPrP1誘導的HSCs活化及肝纖維化形成的調控作用����,分別在體內和體外給予自噬抑制劑LY294002和自噬誘導劑雷帕霉素干預,實驗觀察到減少自噬可以抑制IGFBPrP1誘導的HSCs活化���������,ECM生成減少���������,肝纖維化減弱���;增加自噬可以促進IGFBPrP1誘導的HSCs活化������,ECM生成增多������,肝纖維化加重���。由于LY294002又是PI3K抑制劑����,雷帕霉素是mTOR抑制劑���,研究團隊同時證實PI3K/AKT/mTOR信號通路參與了IGFBPrP1對自噬的調控�����。另外������,通過qRT-PCR���、原位雜交等發現IGFBPrP1也促進了H19的表達�������,給予LY294002和雷帕霉素后發現IGFBPrP1可通過PI3K/AKT/mTOR信號通路促進H19的表達���。最后��������,研究團隊在AdIGFBPrP1轉染JS-1的基礎上,將H19上調或下調���������,發現H19可促進IGFBPrP1誘導的HSCs自噬及活化������,給予LY294002和雷帕霉素后發現PI3K/AKT/mTOR參與了其中的調控������。最后得出的分子調節機制是IGFBPrP1通過PI3K/AKT/mTOR和H19的相互調控促進HSCs自噬及活化���������,進而促進肝纖維化形成������。

圖1小鼠尾靜脈注射腺病毒
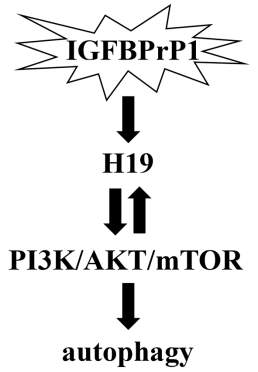
圖2分子調控機制圖
該研究由c7c7.app第一醫院科研中心實驗室劉立新教授團隊完成���,劉立新教授為該論文的通信作者���;c7c7.app2016級博士研究生黃婷娟為該論文的第一作者�����。
該論文以題為“IGFBPrP1 accelerates autophagy and activation of hepatic stellate cells via mutual regulation between H19 and PI3K/AKT/mTOR pathway”發表在Biomedicine & Pharmacotherapy(IF:3.457)雜志上���。

