第一臨床醫學院郭軍紅課題組發現SCN4A基因突變致先天性副肌強直并周期性麻痹的新家系
2019年12月����������,來自c7c7.app第一臨床醫學院的郭軍紅教授及黃珊博士研究生等人的研究“Overlap of periodic paralysis and paramyotonia congenita caused by SCN4A gene mutations two family reports and literature review”在Channels(IF=2.581)上正式刊出。該文章明確了兩個并有間斷肌肉癱瘓及肌強直的離子通道病家系的診斷����,探究周期性麻痹及先天性副肌強直并存的病理機制���,并對國內外相關病例進行了回顧���。
SCN4A基因調控主要存在于骨骼肌表面的鈉離子通道NaV1.4�����,該通道的激活及快速失活影響骨骼肌細胞膜興奮性����,從而進一步參與調控骨骼肌收縮������。SCN4A基因突變可引起骨骼肌興奮性異常,導致先天性副肌強直������、鈉通道性肌強直���、高鉀型周期性麻痹或低鉀型周期性麻痹2型�����。少數患者可出現重疊癥狀������,并存上述兩種診斷��������。
該研究詳細記錄了c7c7.app第一醫院神經內科收治的SCN4A基因突變相關的離子通道病先證者及其家系的基本資料���������、病史����、查體��������,行電生理檢查������、肌肉活檢����������、基因分析���。
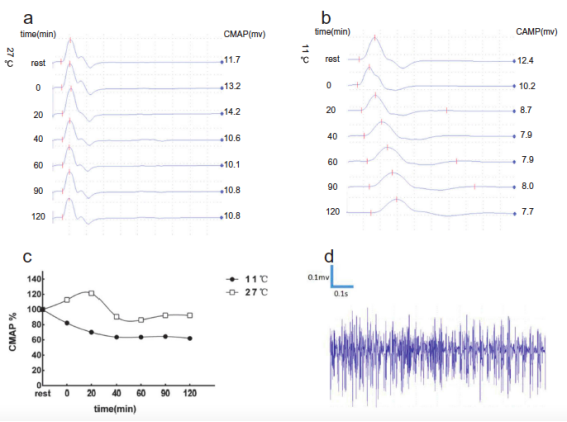
其中一個家系中部分成員基因分析考慮SCN4A基因(T704M)為其致病突變���������,其長時程運動試驗提示患者于常溫所致先天性副肌強直合并高鉀型周期性麻痹�����。
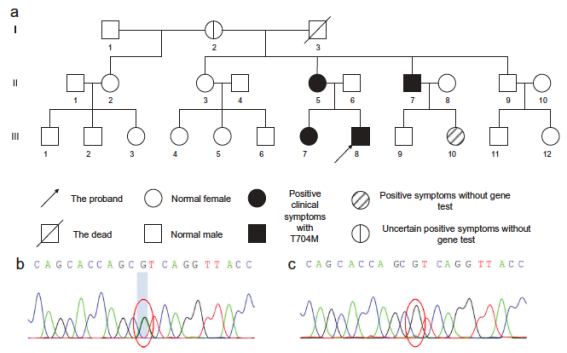

另一個家系中部分成員被診斷為SCN4A基因突變(R1448H)所致先天性副肌強直合并低鉀型周期性麻痹2型��������。此外��������,在此家系中一位具有重疊癥狀的患者為SCN4A基因T704M突變而非R1448H突變�����。

SCN4A基因突變相關的離子通道病肌肉活檢病理變現可見肌纖維中出現管聚集及空洞��������。


該研究提出SCN4A基因突變可致先天性肌強直合并周期性麻痹����,尤其是包括低鉀型周期性麻痹2型��������,這在以往的周期性麻痹病例中十分罕見���。研究同時發現間斷發作的肢體無力及肌強直常發生于不同的時間及溫度���������,為離子通道病后期進一步機制探索提供了思路�����。

